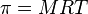chiminigagua

Estudiemos una reacción genérica como la siguiente:
A + B ------ C + D (9)
A medida que la reacción progresa, disminuye el número de moléculas A y B, y aumenta en número de moléculas C y D. Como las sustancias C y D, no reaccionan entre sí, la reacción continua hasta que las moléculas A y B se consumen. Este tipo de reacción se denomina irreversible . Graficando este caso tenemos:
Por ejemplo, la acción del ácido clorhídrico sobre el cinc:
2.HCl + Zn -------- ZnCl2 + H2 (g)
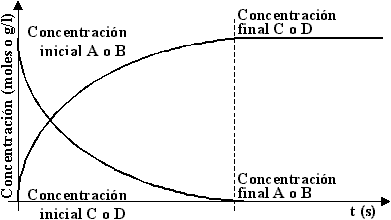
Si en cambio la moléculas C y D pueden reaccionar entre sí, la reacción avanzará hacia la derecha mientras la concentración de las moléculas A y B sea importante, a medida que comiencen a formarse moléculas C y D, la velocidad de reacción disminuirá hasta que la concentración de las moléculas C y D sea tal que la reacción comenzará a desplazarse hacia la izquierda, hasta que se establezca un punto de equilibrio, donde ambas velocidades se equilibran, y en el cual coexistirán moléculas A, B, C y D. Este tipo de reacciones se denominan reversibles, y se representan con flecha de ida y vuelta:
A + B « C + D (10)
El siguiente gráfico representa una reacción reversible:
Un ejemplo de este caso es el siguiente:
CH3-COOH + CH3OH « CH3-CO-O-CH3 + H2O
ácido etanoico + metanol « etanoato de metilo + agua
Cabe aclarar que el concepto de reacción reversible o irreversible no es absoluto.
Hay reacciones reversibles que, según como se realicen, pueden convertirse en irreversibles, por ejemplo, si calentamos carbonato de calcio (CaCO3) en un recipiente cerrado, en un principio, la reacción se desplazará hacia la derecha, produciendo dióxido de carbono y cal (CaO), pero cuando la presión aumente por formación de CO2, la velocidad de reacción hacia la derecha,comenzará a disminuir e irá aumentando la velocidad hacia la izquierda, hasta que quede en equilibrio (reversible).
CaCO3 « CO2 + CaO
En cambio, si se procede en un recipiente abierto, el dióxido de carbono producido, se escapará a la atmósfera, sin dar lugar a la reacción inversa, y continuará hasta la total descomposición del carbonato de calcio (irreversible).
CaCO3 ----- CO2 (g) + CaO
Este es un ejemplo de una reacción reversible o incompleta que, por eliminación de uno de los productos de la reacción, se transforma en irreversible o completa.
La ley de acción de las masas, también se aplica a los sistemas reversibles o en equilibrio. Aplicando la ley a la reacción (10), primero a la que se desplaza a la derecha, tenemos:
v = [A].[B] (11)
Y reemplazando v por kd,que es la constante de la velocidad hacia la derecha:
kd = [A].[B] (12)
Luego, para la reacción hacia la izquierda:
v = [C].[D]
ki = [C].[D] (13)
Dividiendo (13) por (12):
| k = | ki | = | [C].[D] | (14) |
| kd | [A].[B] |
Donde k es la constante de equilibrio.
Analicemos este principio con la reacción (10):
A + B « C + D (10)
1. Los efectos de la concentración: el aumento de las concentraciones de A y B,produce más C y D para contrarrestar el aumento de A y B. Ocurre lo mismo en el caso inverso.
2. Los efectos de la presión: en el caso de que A o B sean gases, el aumento de presión,el sistema reaccionará disminuyendo su volumen para contrarrestar el aumento de presión, con lo cual la reacción se desplazará hacia la derecha.
3. Los efectos de la temperatura: si la reacción entre A y B libera calor (exotérmica),y retiramos las calorías producidas, el sistema reaccionará produciendo más calor para contrarrestar la pérdida, con lo cual la reacción se desplazará hacia la derecha. Si por el contrario, le entregamos calorías, el sistema contrarrestará la modificación desplazándose hacia la izquierda.
El aumento de temperatura retarda los procesos exotérmicos y acelera los endotérmicos.
Ante una reacción reversible (reacción 10), para lograr el máximo rendimiento, es decir que la reacción se desplace lo todo posible hacia la derecha, tenemos las siguientes opciones:
1. Aumentar la concentración de uno de los reactivos, por ejemplo, duplicando A, para contrarrestar este exceso, aumentaran las concentraciones de C y D, desplazando la reacción hacia la derecha.
2.A + B ---- C + D
2. Disminuir la concentración de uno de los productos de reacción,por ejemplo, retirando la mitad de C, para contrarrestar este exceso, disminuirán las concentraciones de A y B,desplazando la reacción hacia la derecha, esto se logra retirando C a medida se produce.
A + B ----- C + 0,5.D
3. Si A y/o B son gases, aumentando la presión (en un recipiente cerrado), logramos desplazar la reacción hacia la derecha.
A(g) + B(g) ----- C + D
4. Si C y/o D son gases, disminuyendo la presión (en un recipiente cerrado), logramos desplazar la reacción hacia la derecha.
A + B ---- C(g) + D(g)
5. Si la reacción entre A y B es exotérmica, una vez alcanzada la energía de activación, conviene refrigerar el sistema para favorecer la producción de C y D.
A + B ------ C + D + cal
6. Si la reacción entre A y B es endotérmica, se deberá entregar calor para alcanzar la energía de activación, y continuar calentando para favorecer la producción de C y D.
A + B ----- C + D - cal
Ejemplo: para la reacción reversible (10), en un recipiente cerrado de 10 litros de capacidad, la constante de equilibrio es k = 0,4; una vez alcanzado el punto de equilibrio, se detectaron las siguientes cantidades: 1,581 moles de A, 1,581 moles de B, 1 moles de C y 1 moles de D. ¿Cómo se puede modificar el sistema para alcanzar un rendimiento de 0,95?
Solución: el rendimiento es
R = masa obtenida real / masa obtenida teórica
Teóricamente:
1.A + 1.B « 1.C + 1.D
6 g + 4 g = 5 g + 5 g
10 g = 10 g
Donde R = 10 g/ 10 g = 1
En nuestro caso:
1,581 mol.A + 1,581 mol.B + 1 mol.C + 1 mol.D
9,487 g + 6,325 g + 5 g + 5 g
15,812 g + 10 g
Donde R1 = 15,812 g/ 10 g = 0,63
La velocidad de reacción para un reactivo en una reaccion quimica en particular está definida intuitivamente como cuán rápido sucede una reacción. Por ejemplo, la oxidación del hierro bajo condiciones atmosféricas es una reacción lenta que puede tomar muchos años,[1] pero la combustión del butano en un fuego es una reacción que sucede en fracciones de segundo.
La cinética química es la parte de la fisicoquímica que estudia las velocidades de reacción, la dinámica química estudia los orígenes de las diferentes velocidades de las reacciones. El concepto de cinética química se aplica en muchas disciplinas, tales como la ingeniería química, enzimología e ingeniería ambiental.
Considérese una reacción química típica:
aA + bB → pP + qQLas letras minúsculas (a, b, p, y q) representan los coeficientes estequiométricos, mientras que las letras mayúsculas representan a los reactante (A y B) y los productos (P y Q). De acuerdo a la definición del Libro Dorado de la IUPAC[2] la velocidad de reacción v (también r o R) de una reacción química que se da en un sistema cerrado bajo condiciones de volumen constante, sin que haya acumulación de intermediarios de reacción, está definida por:
![v = - frac{1}{a} frac{d[A]}{dt} = - frac{1}{b} frac{d[B]}{dt} = frac{1}{p} frac{d[P]}{dt} = frac{1}{q} frac{d[Q]}{dt}](https://mastersofchemistry.blogia.com/upload/externo-09bbe9b2625261d27e830cfb1b65f270.png)
(NOTA:La velocidad de reacción es siempre positiva. El signo '-' está presente en los términos que involucran a los reactantes porque la concentración de reactante disminuye en el tiempo.) La IUPAC[2] recomienda que la unidad de tiempo siempre deba ser el segundo. En tal caso, la velocidad de reacción difiere de la velocidad de aumento de la concentración de un producto P por un factor constante (el recíproco de su número estequiométrico) y por un reactante A por menos el recíproco del número estequiométrico. Generalmente, la velocidad de reacción tiene las unidades dm−3·s−1.
Es importante tener en cuenta que la definición previa es válida sólo para una sola reacción, en un sistema cerrado de volumen constante. Esta suposición muy frecuentemente implícita debe ser explicitada, de lo contrario la definición es incorrecta: si se agrega agua a un recipiente conteniendo agua salada, la concentración de la sal disminuye, aunque no haya reacción química.
La IUPAC[2] recomienda el uso de los términos velocidad de aparición y velocidad de desaparición para los productos y reactantes, respectivamente.
Las velocidades de reacción también pueden ser definidas usando una base diferente al volumen del reactor. Cuando se usa un catalizador, la velocidad de reacción puede ser expresada en base al peso del catalizador (mol g−1 s−1) o área de la superficie del mismo (mol m−2 s−1). Si se toma como base un sitio específico de un catalizador que puede ser contado rigurosamente por un método específico, la velocidad puede ser expresada en unidades de s−1, por lo que se le denomina frecuencia de cambio, o de conversión.~~
La velocidad de reacción puede ser independiente de la temperatura (no-Arrhenius) o disminuir con el aumento de la temperatura (anti-Arrhenius). Las reacciones sin una barrera de activación (por ejemplo, algunas reacciones de radicales) tienden a tener una dependencia de la temperatura de tipo anti Arrhenius: la constante de velocidad disminuye al aumentar la temperatura.
Todos los factores que afectan una velocidad de reacción, excepto para la concentración y el orden de reacción, son tomados en cuenta en la ecuación de velocidad de la reacción.
Para una reacción química n A + m B → C + D, la ecuación de velocidad o ley de reacción es una expresión matemática usada en cinética química que relaciona la velocidad de una reacción con la concentración de cada reactante. Es del tipo:
![,r = k(T)[A]^{n'}[B]^{m'}](https://mastersofchemistry.blogia.com/upload/externo-2a2a3ae3a7eadd60a302525a21984cf0.png)
En esta ecuación, k(T) es el coeficiente cinético de reacción o constante de velocidad, aunque no es realmente una constante, debido a que incluye todos los parámetros que afectan la velocidad de reacción, excepto la concentración, que es explícitamente tomada en cuenta. De todos los parámetros descritos anteriormente, normalmente la temperatura es el más importante. Los exponentens n' y m' son denominados órdenes y dependen del mecanismo de reacción.
La estequiometría, molecularidad (el número real de moléculas que colisionan) y el orden de reacción sólo coinciden necesariamente en las reacciones elementales, esto es en las reacciones que proceden en un solo paso. La ecuación de reacción para reacciones elementales coincide con el proceso que tiene lugar a nivel atómico, donde n moléculas del tipo A colisionan con m moléculas del tipo B (n más m es la molecularidad).
Para gases, la ley de velocidad puede ser expresada también en unidades de presión, usando la ley de gases ideales. Al combinar la ley de velocidad con un balance de masa para el sistema en el que sucede la reacción, puede derivarse una expresión para la velocidad de cambio en la concentración. Para un sistema cerrado con un volumen constante, tal expresión puede verse como:
![frac{d[C]}{dt} = k(T)[A]^{n'}[B]^{m'}](https://mastersofchemistry.blogia.com/upload/externo-e1e3c094500bc03e60f0b550b05a193f.png)
Cada coeficiente de velocidad de reacción k tiene una dependencia de la temperatura, que es dada usualmente por la ecuación de Arrhenius:
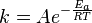
Ea es la energía de activación y R es la constante universal de los gases. Dado que a la temperatura T, las moléculas tienen energías dadas por una distribución de Boltzmann, se puede esperar que el número de colisiones con energía mayor que Ea sea proporcional a 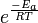 . A es el factor pre-exponencial o factor de frecuencia o factor de Arrhenius.
. A es el factor pre-exponencial o factor de frecuencia o factor de Arrhenius.
Los valores de A y Ea son dependientes de la reacción. También es posible ecuaciones más complejas, que describen la dependencia de la temperatura de otras constantes de velocidad, que no siguen este esquema.
La dependencia de la presión de la constante de velocidad para reacciones en fase condensada (por ejemplo, cuando los reactantes son sólidos o líquidos) es, por lo general, suficientemente débil en el rango de las presiones que se encuentran normalmente en la industria, por lo que suele ser despreciada en la práctica. La dependencia de la presión está asociada con el volumen de activación. Para la reacción siguiente que procede a través de un estado de activación complejo:
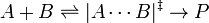
El volumen de activación, 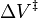 , es:
, es:
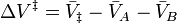
donde 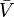 denota los volúmenes molares parciales de los reactantes y productos, y
denota los volúmenes molares parciales de los reactantes y productos, y 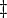 indica el complejo del estado de activación. Para la reacción anterior, puede esperarse que el cambio de la constante de velocidad de reacción (basada en fracción molar o concentración molar) con la presión, a temperatura constante, sea:
indica el complejo del estado de activación. Para la reacción anterior, puede esperarse que el cambio de la constante de velocidad de reacción (basada en fracción molar o concentración molar) con la presión, a temperatura constante, sea:
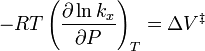
En la práctica, la materia puede ser complicada, debido a que los volúmenes molares parciales y el volumen de activación pueden ser también una función de la presión.
Las reacciones también pueden tener su velocidad incrementada o disminuida con la presión, dependiente del valor de . Como ejemplo de la posible magnitud del efecto de la presión, se encontró que algunas reacciones orgánicas doblaban la velocidad de reacción cuando la presión era incrementada desde la atmosférica (0,1 MPa) hasta 50 MPa (lo que da =-0.025 L/mol).[4]
Para la reacción
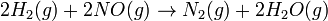
La ecuación de velocidad es:
![r = k [H_2]^1[NO]^2 ,](https://mastersofchemistry.blogia.com/upload/externo-43a8874e49982ab421cbafc98634cf30.png)
La ecuación de velocidad no refiere simplemente los coeficientes estequiométricos de los reactantes en la reacción global: es de primer orden en H2, aunque el coeficiente estequiométrico es 2, y es de segundo orden en NO.
En cinética química, se suele proponer que la reacción global ocurre a través de una serie discreta de pasos elementales. No todos los pasos afectan la velocidad de reacción; normalmente sólo el paso elemental más lento es el que afecta la velocidad de reacción. Por ejemplo, en:
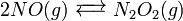 (equilibrio rápido)
(equilibrio rápido)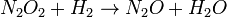 (lenta)
(lenta)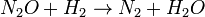 (rápida)
(rápida)Las reacciones 1 y 3 son muy rápidas comparadas con la segunda, por lo que es la reacción más lenta la que es reflejada en la ecuación de velocidad. El paso lento es considerado la etapa limitante de la velocidad. Los órdenes de la ecuación de velocidad son aquellos de la etapa limitante.
En química se llaman propiedades coligativas a aquellas propiedades de una disolución que dependen únicamente de la concentración (generalmente expresada comoconcentración molar, es decir, de la cantidad de partículas de soluto por partículas totales, y no de la naturaleza o tipo de soluto.
Están estrechamente relacionadas con la presión de vapor, que es la presión que ejerce la fase de vapor sobre la fase líquida, cuando el líquido se encuentra en un recipiente cerrado. La presión de vapor depende del solvente y de la temperatura a la cual sea medida (a mayor temperatura, mayor presión de vapor). Se mide cuando el sistema llega al equilibrio dinámico, es decir, cuando la cantidad de moléculas de vapor que vuelven a la fase líquida es igual a las moléculas que se transforman en vapor.
Cuando se prepara una solución con un solvente y un soluto no volátil (que se transformará en gas) y se mide su presión, al compararla con la presión de vapor de su solvente puro (medidas a la misma temperatura), se observa que la de la solución es menor que la del solvente. Esto es consecuencia de la presencia del soluto no volátil.
A su vez, cuando se las comparan las presiones de vapor de dos soluciones de igual composición y diferente concentración, aquella solución más concentrada tiene menor presión de vapor. El descenso de ésta se produce por dos razones: por probabilidad, pues es menos probable que existan moléculas de disolvente en el límite de cambio, y por cohesión, pues las moléculas de soluto atraen a las de disolvente por lo que cuesta más el cambio.
La presión de vapor de un disolvente desciende cuando se le añade un soluto no volátil.
Este efecto es el resultado de dos factores:
El soluto obstaculiza la formación de cristales sólidos, por ejemplo el líquido anticongelante de los motores de los automóviles tiene una base de agua pura a presión atmosférica se congelaría a 0°C dentro de las tuberías y no resultaría útil en lugares fríos. Para evitarlo se le agregan ciertas sustancias químicas que hacen descender su punto de congelación.
ΔTf = Kf · mPara enfriar algo rápidamente se hace una mezcla de hielo con sal o, si tiene precaución, alcohol. El punto de congelación bajará y el hielo se derretirá rápidamente. Pese a aparentar haberse perdido el frío, la mezcla formada estará en realidad a unos cuantos grados bajo cero y será mucho más efectiva para enfriar que los cubos de hielo sólidos. Es una consecuencia del descenso de la presión de vapor.
El agua se congela a partir de los 0 °C, mientras que una solución formada por agua y sal se congelará a menor temperatura (de ahí que se utilice sal para fundir nieve o hielo con mayor facilidad)
La congelación es la aplicación más drástica del frío
• Temperatura del alimento < punto de congelación
• Temperaturas de conservación mas o menos -20 °C
• Disminuye la actividad del agua (forma de hielo)
• No hay desarrollo microbiano, pero no destruye todas las bacterias
• Limita la acción de la mayoría de las reacciones químicas y enzimáticas
• Aumento de la vida útil de los alimentos
• Se mantienen las características organolépticas y valor nutritivo si el proceso de congelación y almacenamiento son los adecuados
•La Congelación es el mejor método para conservación a largo plazo
•La Congelación y almacenamiento realizados correctamente permiten la no variación de propiedades organolépticas y nutritivas y una vida útil elevada. [1]
Al agregar moléculas o iones a un solvente puro la temperatura en el que éste entra en ebullición es más alto. Por ejemplo, el agua pura a presión atmosférica ebulle a 100 °C, pero si se disuelve algo en ella el punto de ebullición sube algunos grados centígrados.
ΔTb = Kb · mCuando un mol de una sal se disuelve en solución, el efecto del aumento del punto de ebullición es aún mayor, pues la sal hará un efecto tal que será el total de las partes que se disuelven. Por ejemplo, el NaCl será disuelto en un mol de sodio y un mol de cloro, un total de dos moles en solución.
El punto de ebullición es la temperatura a la cual la presión de vapor de un solvente o solución iguala la presión externa y comienza a observarse las moléculas de líquido transformarse en gas. Por ejemplo, a presión externa de 1 atm, el agua hierve a 100 °C, mientras que para una solución acuosa de algo a 100 °C las presiones externas y de vapor no se han igualado y por ende no se observa el cambio a estado gaseoso. Cuando la presión de vapor iguale la presión externa la temperatura de la solución será mayor que 100 °C y, consecuentemente, se comprueba que su punto de ebullición es, efectivamente, mayor que el punto de ebullición de su solvente puro (agua) medido a una misma presión externa.
La ósmosis es la tendencia que tienen los solventes a ir desde zonas de mayor concentración hacia zonas de menor concentración de partículas. El efecto puede pensarse como una tendencia de los solventes a "diluir". Es el pasaje espontáneo de solvente desde una solución más diluida (menos concentrada) hacia una solución menos diluida (más concentrada), cuando se hallan separadas por una membrana semipermeable.
(también: π = (nRT) / V)
Teniendo en cuenta que n/V representa la molaridad (M) de la solución obtenemos:
Al igual que en la ley de los gases ideales, la presión osmótica no depende de la carga de las partículas.
Observación: Se utiliza la unidad de Molaridad (M) para expresar la concentración ya que el fenómeno de ósmosis ocurre a temperatura constante (de esto se deduce que las unidades de concentración para el ascenso ebulloscópico y el descenso crioscópico estén dadas en molalidad (m), ya que este tipo de expresión no varía con la temperatura).
El experimento más típico para observar el fenómeno de ósmosis es el siguiente:
Las membranas celulares son semipermeables. La observación al microscopio de células que previamente han estado sumergidas en soluciones de sal común o azúcar, permite constatar el efecto de la entrada de agua (turgencia) o la pérdida de agua (plasmólisis) en función de que el medio exterior sea hipertónico o hipotónico respecto al medio interno celular
La cromatografía es un método físico de separación para la caracterización de mezclas complejas, la cual tiene aplicación en todas las ramas de la ciencia y la física. Es un conjunto de técnicas basadas en el principio de retención selectiva, cuyo objetivo es separar los distintos componentes de una mezcla, permitiendo identificar y determinar las cantidades de dichos componentes.
Las técnicas cromatográficas[1] son muy variadas, pero en todas ellas hay una fase móvil que consiste en un fluido (gas, líquido o fluido supercrítico) que arrastra a la muestra a través de una fase estacionaria que se trata de un sólido o un líquido fijado en un sólido. Los componentes de la mezcla interaccionan en distinta forma con la fase estacionaria. De este modo, los componentes atraviesan la fase estacionaria a distintas velocidades y se van separando. Después de que los componentes hayan pasado por la fase estacionaria, separándose, pasan por un detector que genera una señal que puede depender de la concentración y del tipo de compuesto.
Diferencias sutiles en el coeficiente de partición de los compuestos da como resultado una retención diferencial sobre la fase estacionaria y por tanto una separación efectiva en función de los tiempos de retención de cada componente de la mezcla.
La cromatografía puede cumplir dos funciones básicas que no se excluyen mutuamente:
| Tipos | Fase movida | Fase estacionaria |
| Cromatografía en papel | Líquido | Líquido ( moléculas de agua contenidas en la celulosa del papel ) |
| Cromatografía en capa fina | Líquido | Sólido |
| Cromatografía de gases | Gas | Sólido o líquido |
| Cromatografía líquida en fase inversa | Líquido (polar) | Sólido o líquido (menos polar) |
| Cromatografía líquida en fase normal | Líquido (menos polar) | Sólido o líquido (polar) |
| Cromatografía líquida de intercambio iónico | Líquido (polar) | Sólido |
| Cromatografía líquida de exclusión | Líquido | Sólido |
| Cromatografía líquida de adsorción | Líquido | Sólido |
| Cromatografía de fluidos supercríticos | Líquido | Sólido |
Las distintas técnicas cromatográficas se pueden dividir según cómo esté dispuesta la fase estacionaria:
La cromatografía de gases es útil para gases o para compuestos relativamente volátiles, lo que incluye a numerosos compuestos orgánicos. En el caso de compuestos no volátiles se recurre a procesos denominados de "derivatización", a fin de convertirlos en otros compuestos que se volatilizen en las condiciones de análisis.
Dentro de la cromatografía líquida destaca la cromatografía líquida de alta resolución (HPLC, del inglés High Performance Liquid Chromatography), que es la técnica cromatográfica más empleada en la actualidad, normalmente en su modalidad de fase reversa, en la que la fase estacionaria tiene carácter no polar, y la fase móvil posee carácter polar (generalmente agua o mezclas con elevada proporción de la misma, o de otros disolvente polares, como por ejemplo metanol). El nombre de "reversa" viene dado porque tradicionalmente la fase estacionaria estaba compuesta de sílice o alúmina, de carácter polar, y por tanto la fase móvil era un disolvente orgánico poco polar.
Una serie eluotrópica, es un rango de sustancias de diferentes polaridades que actúan como fase móvil y que permiten observar un mejor desplazamiento sobre una fase estacionaria.
El botánico ruso Mijaíl Tswett[4] (Mikhail Semenovich Tswett, 1872-1919) empleó por primera vez en 1906 el término "cromatografía" (que proviene del griego χρομα y γραφω que significan respectivamente chroma "color" y graphos "escribir").

A comienzos del año 1903, Mijail Tsvet usó columnas de adsorción de líquidos para separar pigmentos vegetales (por ejemplo, clorofilas). Las disoluciones se hacían pasar a través de una columna de vidrio rellena de carbonato de calcio, que finamente dividido de un material poroso que interacciona de forma diferente con los componentes de la mezcla, de forma que éstos se separaban en distintas bandas coloreadas a lo largo de la columna.
Los primeros equipos de cromatografía de gases aparecieron en el mercado a mediados del siglo XX. A su vez, la cromatografía líquida de alta resolución (HPLC) comenzó a desarrollarse en los años 1960, aumentando su importancia en las décadas siguientes, hasta convertirse en la técnica cromatográfica más empleada. Sin embargo esto se irá modificando con el paso de los años.

 En el eje X se representa el tiempo de retención, y en el eje Y una señal (obtenida, por ejemplo, a partir de un espectrofotómetro, un espectrómetro de masas o cualquier otro de los diversos detectores) correspondiente a la respuesta creada por los diferentes analitos existentes en la muestra. En el caso de un sistema óptimo, la señal es proporcional a la concentración del analito específico separado.
En el eje X se representa el tiempo de retención, y en el eje Y una señal (obtenida, por ejemplo, a partir de un espectrofotómetro, un espectrómetro de masas o cualquier otro de los diversos detectores) correspondiente a la respuesta creada por los diferentes analitos existentes en la muestra. En el caso de un sistema óptimo, la señal es proporcional a la concentración del analito específico separado. 
La destilación es la operación de separar, mediante vaporización y condensación, los diferentes componentes líquidos, sólidos disueltos en líquidos o gases licuados de una mezcla, aprovechando los diferentes puntos de ebullición (temperaturas de ebullición) de cada una de las sustancias ya que el punto de ebullición es una propiedad intensiva de cada sustancia, es decir, no varia en función de la masa o el volumen, aunque sí en función de la presión.
El aparato utilizado para la destilación en el laboratorio es el '''alambique'''. Consta de un recipiente donde se almacena la mezcla a la que se le aplica calor, un condensador donde se enfrían los vapores generados, llevándolos de nuevo al estado líquido y un recipiente donde se almacena el líquido concentrado.
En la industria química se utiliza la destilación para la separación de mezclas simples o complejas. Una forma de clasificar la destilación puede ser la de que sea discontinua o continua.
En el esquema de la derecha puede observarse un aparato de destilación simple básico:

La destilación fraccionada es una variante de la destilación simple que se emplea principalmente cuando es necesario separar líquidos con punto de ebullición cercanos.
La principal diferencia que tiene con la destilación simple es el uso de una columna de fraccionamiento. Ésta permite un mayor contacto entre los vapores que ascienden con el líquido condensado que desciende, por la utilización de diferentes "platos". Esto facilita el intercambio de calor entre los vapores (que ceden) y los líquidos (que reciben). Ese intercambio produce un intercambio de masa, donde los líquidos con menor punto de ebullición se convierten en vapor, y los vapores con mayor punto de ebullición pasan al estado líquido.
La destilación a vacío es la operación complementaria de destilación del crudo procesado en la unidad de destilación atmosférica, que no se vaporiza y sale por la parte inferior de la columna de destilación atmosférica. El vaporizado de todo el crudo a la presión atmosférica necesitaría elevar la temperatura por encima del umbral de descomposición química y eso, en esta fase del refino de petróleo, es indeseable.
El residuo atmosférico o crudo reducido procedente del fondo de la columna de destilación atmosférica, se bombea a la unidad de destilación a vacío, se calienta generalmente en un horno a una temperatura inferior a los 400 °C, similar a la temperatura que se alcanza en la fase de destilación atmosférica, y se introduce en la columna de destilación. Esta columna trabaja a vacío, con una presión absoluta de unos 20 mm de Hg, por lo que se vuelve a producir una vaporización de productos por efecto de la disminución de la presión, pudiendo extraerle más productos ligeros sin descomponer su estructura molecular.
En la unidad de vacío se obtienen solo tres tipos de productos:
Los dos primeros, GOL y GOP, se utilizan como alimentación a la unidad de craqueo catalítico después de desulfurarse en una unidad de hidrodesulfuración (HDS).
El producto del fondo, residuo de vacío, se utiliza principalmente para alimentar a unidades de craqueo térmico, donde se vuelven a producir más productos ligeros y el fondo se dedica a producir fuel oil, o para alimentar a la unidad de producción de coque. Dependiendo de la naturaleza del crudo el residuo de vacío puede ser materia prima para producir asfaltos.
En química, la destilación azeotrópica es una de las técnicas usadas para romper un azeótropo en la destilación. Una de las destilaciones más comunes con un azeótropo es la de la mezcla etanol-agua. Usando técnicas normales de destilación, el etanol solo puede purificarse a aproximadamente el 95%.
Una vez se encuentra en una concentración de 95/5% etanol/agua, los coeficientes de actividad del agua y del etanol son iguales, entonces la concentración del vapor de la mezcla también es de 95/5% etanol-agua, por lo tanto destilar de nuevo no es efectivo. Algunos usos requieren concentraciones de alcohol mayores, por ejemplo cuando se usa como aditivo para la gasolina. Por lo tanto el azeótropo 95/5% debe romperse para lograr una mayor concentración.
En uno de los métodos se adiciona un material agente de separación. Por ejemplo, la adición de benceno a la mezcla cambia la interacción molecular y elimina el azeótropo. La desventaja, es la necesidad de otra separación para retirar el benceno. Otro método, la variación de presión en la destilación, se basa en el hecho de que un azeótropo depende de la presión y también que no es un rango de concentraciones que no pueden ser destiladas, sino el punto en el que los coeficientes de actividad se cruzan. Si el azeótropo se salta, la destilación puede continuar.
Para saltar el azeótropo, el punto del azeótropo puede moverse cambiando la presión. Comúnmente, la presión se fija de forma tal que el azeótropo quede cerca del 100% de concentración, para el caso del etanol, éste se puede ubicar en el 97%. El etanol puede destilarse entonces hasta el 97%. Actualmente se destila a un poco menos del 95,5%. El alcohol al 95,5% se envía a una columna de destilación que está a una presión diferente, se lleva el azeótropo a una concentración menor, tal vez al 93%. Ya que la mezcla está por encima de la concentración azeotrópica actual, la destilación no se “pegará” en este punto y el etanol se podrá destilar a cualquier concentración necesaria.
Para lograr la concentración requerida para que el etanol sirva como aditivo de la gasolina normalmente se usan tamices moleculares en la concentración azeotrópica. El etanol se destila hasta el 95%, luego se hace pasar por un tamiz molecular que absorba el agua de la mezcla, ya se tiene entonces etanol por encima del 95% de concentración, que permite destilaciones posteriores. Luego el tamiz se calienta para eliminar el agua y puede reutilizarse.
En la destilación por arrastre de vapor de agua se lleva a cabo la vaporización selectiva del componente volátil de una mezcla formada por éste y otros "no volátiles". Lo anterior se logra por medio de la inyección de vapor de agua directamente en el interior de la mezcla, denominándose este "vapor de arrastre", pero en realidad su función no es la de "arrastrar" el componente volátil, sino condensarse en el matraz formando otra fase inmiscible que cederá su calor latente a la mezcla a destilar para lograr su evaporación. En este caso se tendrán la presencia de dos fases insolubles a lo largo de la destilación (orgánica y acuosa), por lo tanto, cada líquido se comportará como si el otro no estuviera presente. Es decir, cada uno de ellos ejercerá su propia presión de vapor y corresponderá a la de un líquido puro a una temperatura de referencia.
La condición más importante para que este tipo de destilación pueda ser aplicado es que tanto el componente volátil como la impureza sean insolubles en agua ya que el producto destilado volátil formará dos capas al condensarse, lo cual permitirá la separación del producto y del agua fácilmente.
Como se mencionó anteriormente, la presión total del sistema será la suma de las presiones de vapor de los componentes de la mezcla orgánica y del agua, sin embargo, si la mezcla a destilar es un hidrocarburo con algún aceite, la presión de vapor del aceite al ser muy pequeña se considera despreciable a efectos del cálculo:
P = Pa° + Pb°
Donde:
Por otra parte, el punto de ebullición de cualquier sistema se alcanza a la temperatura a la cual la presión total del sistema es igual a la presión del confinamiento. Y como los dos líquidos juntos alcanzan una presión dada, más rápidamente que cualquiera de ellos solos, la mezcla hervirá a una temperatura más baja que cualquiera de los componentes puros. En la destilación por arrastre es posible utilizar gas inerte para el arrastre. Sin embargo, el empleo de vapores o gases diferentes al agua implica problemas adicionales en la condensación y recuperación del destilado o gas.
El comportamiento que tendrá la temperatura a lo largo de la destilación será constante, ya que no existen cambios en la presión de vapor o en la composición de los vapores de la mezcla, es decir que el punto de ebullición permanecerá constante mientras ambos líquidos estén presentes en la fase líquida. En el momento que uno de los líquidos se elimine por la propia ebullición de la mezcla, la temperatura ascenderá bruscamente.
Si en mezcla binaria designamos por na y nb a las fracciones molares de los dos líquidos en la fase vapor, tendremos:
na y nb son el número de moles de A y B en cualquier volúmen dado de vapor, por lo tanto:
y como la relación de las presiones de vapor a una "T" dada es constante, la relación na/nb, debe ser constante también. Es decir, la composición del vapor es siempre constante en tanto que ambos líquidos estén presentes.
Además como: na = wa/Ma y nb= wb/Mb
donde: wa y wb son los pesos en un volúmen dado y Ma, Mb son los pesos moleculares de A y B respectivamente. La ecuación se transforma en:
Pa° = na = waMb Pb° nb wbMa O bien: wa = MaPa° wb MbPb°
Esta última ecuación relaciona directamente los pesos moleculares de los dos componentes destilados, en una mezcla binaria de líquidos. Por lo tanto, la destilación por arrastre con vapor de agua, en sistemas de líquidos inmisibles en ésta se llega a utilizar para determinar los pesos moleculares aproximados de los productos o sustancias relacionadas.
Es necesario establecer que existe una gran diferencia entre una destilación por arrastre y una simple, ya que en la primera no se presenta un equilibrio de fases líquido-vapor entre los dos componentes a destilar como se da en la destilación simple, por lo tanto no es posible realizar diagramas de equilibrio ya que en el vapor nunca estará presente el componente "no volátil" mientras esté destilando el volátil. Además de que en la destilación por arrastre de vapor el destilado obtenido será puro en relación al componente no volátil (aunque requiera de un decantación para ser separado del agua), algo que no sucede en la destilación simple donde el destilado sigue presentando ambos componentes aunque más enriquecido en alguno de ellos. Además si este tipo de mezclas con aceites de alto peso molecular fueran destiladas sin la adición del vapor se requeriría de gran cantidad de energía para calentarla y emplearía mayor tiempo, pudiéndose descomponer si se trata de un aceite esencial.
Cuando existen dos o más compuestos en una mezcla que tienen puntos de ebullición relativamente cercanos, es decir, volatilidad relativa menor a 1 y que forma una mezcla no ideal es necesario considerar otras alternativas más económicas a la destilación convencional, como son:
Estas técnicas no son ventajosas en todos los casos y las reglas de análisis y diseño pueden no ser generalizables a todos los sistemas, por lo que cada mezcla debe ser analizada cuidadosamente para encontrar las mejores condiciones de trabajo.
Mezclas heterogéneas.
Una mezcla heterogénea o sistema material heterogéneo es un sistema material formado por varias sustancias en el que su composición, estructura o propiedades no se mantienen en cualquier punto de su masa, pudiéndose percibir límites de separación entre regiones diversas.
Algunas veces no resulta fácil comprobar si un sistema material es una mezcla heterogénea. Puede que a simple vista encontremos cierta apariencia de regularidad y de uniformidad pero un análisis más cuidadoso puede advertirnos ciertas diferencias. La mayoría de los materiales de uso habitual son heterogéneos y sólo algunos pueden considerarse realmente homogéneos.
La forma de separar las sustancias que forman una mezcla utilizará algunas de las propiedades de las mismas, propiedades que sean diferentes entre las sustancias que la forman.
Así, por ejemplo, si una de las sustancias es atraída por los imanes, utilizaremos un imán para separarla del resto de sustancias que forman la mezcla heterogénea.
Si la diferencia es el tamaño de las sustancias que constituyen la mezcla heterogénea:
Si todas las sustancias son sólidas y el tamaño es muy diferente, podemos utilizar una criba para dejar pasar las más pequeñas y dejar en la criba las más grandes.
Para separar sólidos de líquidos podemos:
Si la densidad es muy diferente, sólido más denso que el líquido, podemos dejar sedimentar (dejar que con el tiempo el sólido se deposite en el fondo) y coger la parte superior (líquido) volcando ligeramente el vaso que lo contenga (decantación). Previamente podríamos haber centrifugado el sistema para que el sólido quedara más apelmazado en el fondo del vaso (centrifugación) y al volcar pudiéramos separa mayor cantidad de líquido.
También podríamos utilizar la filtración: tipo de criba en la que el tamaño de los agujeros es sumamente pequeño.
Si queremos separar líquidos de diferente densidad y no miscibles (no se disuelven unos en otros) podemos utilizar el proceso anterior de decantación:
Volcando ligeramente el vaso que contiene los líquidos la fase superior la podemos trasvasar a otro recipiente y quedarnos en el vaso con la fase inferior (la de menos densidad).
Utilizando un embudo de decantación. El embudo de decantación tiene una salida en la parte inferior con una llave de forma que cuando se encuentren bien delimitadas las fases podemos ir separándolas abriendo la llave y separando la parte inferior. Con el embudo de decantación podemos lograr separaciones de líquidos por el método de decantación con una precisión mucho mayor que el simple volcado del vaso.
Resumen de métodos:
Sólido de sólido:
Imantación si uno de ellos es atraído por imanes.
Manual si el tamaño lo permite.
Criba si son de diferentes tamaños.
Sólido de líquido:
Sedimentación seguido de decantación.
Sedimentación y centrifugación, seguido de decantación.
Filtración.
Líquido de líquido, no miscibles:
Decantación normal.
Decantación, utilizando el embudo de decantación.
No hay que olvidar que el utilizar uno u otro método depende de las características de las sustancias a separar y de qué interesa obtener de forma más pura.
Mezclas homogéneas o disoluciones.
Una mezcla homogénea es un sistema material homogéneo formado por varias sustancias. Las mezclas homogéneas se llaman disoluciones.
Nos encontramos con disoluciones de sustancias que se encuentran cualquier estado de agregación con otras sustancias que se encuentran en el mismo estado de agregación o en otro diferentes.
En una disolución denominamos disolvente a la sustancia de la mezcla que se encuentra en mayor proporción. Denominamos soluto a la sustancia o sustancias que se encuentran en menor proporción.
Soluto | Disolvente | Comentarios y ejemplos |
| Sólido | Sólido | Son las aleaciones. |
| Líquido | Amalgamas. | |
| Gas | El más habitual es el hidrógeno en determinados metales. | |
| Sólido | Líquido | Son las disoluciones más habituales, las que se suelen utilizar en química. |
| Líquido | Cuando los líquidos se disuelven uno en el otro, por ejemplo alcohol en agua. | |
| Gas | Siempre se suele disolver algo de gas en los líquidos. Por ejemplo, el aire disuelto en el agua, las bebidas gaseosas, etc. | |
| Sólido | Gas | Humo. |
| Líquido | Niebla. | |
| Gas | Por ejemplo, el más habitual es el aire. |
Como se ha dicho anteriormente, los métodos de separación se basan en diferencias entre las propiedades físicas de los componentes de una mezcla. Para las disoluciones, los métodos más habituales son:
Destilación. Este método consiste en separar los componentes de las mezclas basándose en lo volátiles que sean las sustancias que forman la mezcla se utilizan los destiladores. Una sustancia de punto de ebullición bajo se considera “volátil” en relación con las otras sustancias de puntos de ebullición mayor. Hay varios tipos de destilación, la más sencilla es la destilación simple en la que el proceso se lleva a cabo por medio de una sola etapa, es decir, que se evapora el líquido de punto de ebullición más bajo (calentando la mezcla) y se condensa por medio de un refrigerante.
Evaporación y cristalización:
La evaporación consiste en eliminar el disolvente líquido, quedándonos con el soluto. Para favorecer la evaporación podemos calentar la mezcla o dejar que ocurra lentamente.
La cristalización es el depósito del sólido disuelto en el líquido por alguno de los siguientes motivos:
Por enfriamiento, habitualmente se disuelven mejor los sólidos en los líquidos la aumentar la temperatura. Si nosotros enfriamos deberá tener menos sólido disuelto en el líquido, el sólido que sobra acabará depositándose en el fondo del recipiente (cristalización).
Por evaporación, al disminuir la cantidad de disolvente deberá tener menos sólido disuelto, el que vaya sobrando a medida que se evapore el líquido se depositará en el fondo del recipiente (cristalización).
Extracción. Consiste en separar varios solutos disueltos en un disolvente. Se utiliza la diferencia de solubilidad (ver apartado cuarto) de cada soluto en diferentes disolventes. Se añade un disolvente inmiscible (que no se disuelve) con el disolvente de la mezcla, los solutos se distribuyen entre los dos disolventes: alguno de los solutos será más soluble en el primer disolvente y otro de los solutos en el segundo disolvente. Posteriormente las dos fases se separan como mezclas heterogéneas, por decantación en este caso.
Cromatografía. Las sustancias a separar se distribuyen entre dos fases según la tendencia que tengan a estar más en una de las fases o en la otra. Una fase es la denominada móvil, la que avanza, y la otra es la fase fija. Los más solubles o que retiene mejor la fase fija retrasan su avance y, de esta forma, se separan de los que retiene mejor la fase móvil.
Sustancias puras.
Son sistemas materiales homogéneos formados de un solo tipo de sustancia. Pueden ser de dos tipos:
Simples o elementos. Son sustancias de composición simple y que no pueden descomponerse en otras más sencillas por métodos químicos ordinarios. Son los elementos químicos.
Compuestos. Son sustancias formadas por la unión química, o combinación, de dos o más elementos en proporciones fijas, siendo las propiedades del compuesto diferentes de las de sus elementos constituyentes. Los compuestos se pueden descomponer en los elementos que los constituyen por métodos químicos habituales.
¿Cómo diferenciar compuestos de disoluciones (mezclas homogéneas)?
Compuesto | Disolución (mezcla homogénea) |
Los constituyentes del compuesto (elementos) se encuentran en proporciones fijas. | Los constituyentes de la mezcla pueden estar en cualquier proporción. |
Si al calentar o enfriar alcanzamos la temperatura de fusión o de ebullición, esta se mantiene estable mientras no cambie el estado de agregación de la sustancia. | Si al calentar o enfriar alcanzamos la temperatura de fusión o de ebullición de uno de los componentes de la mezcla; esta temperatura se estabiliza algo pero no se mantiene invariable porque sólo está cambiando el estado de una de las sustancias que forman la mezcla, la otra u otras siguen aumentando su temperatura. |
Los compuestos se pueden separar en los elementos que lo constituyen por medios químicos. | Las mezclas se pueden separar en las sustancias que la constituyen por medios físicos sencillos. |
Sistemas Homogéneos: si observamos las propiedades intensivas de una muestra de agua pura contenida en un recipiente (Punto de fusión, punto de ebullición, densidad, etc.), veremos que ellas permanecen constantes para cualquier porción de agua que se considere. El agua es el único componente del sistema.
Si ahora consideramos un sistema formado por el agua a la que le hemos agregado una pequeña cantidad de azúcar -sistema formado por dos componentes: agua y azúcar-, podemos observar y comprobar que las propiedades intensivas en este caso son iguales en todos los puntos de su masa.
Decimos entonces que, el sistema de un componente,agua pur, y el sistema de dos componentes, agua y azúcar, constituyen sistemas homogéneos.
Definimos sistema homogéneo: a aquel que presenta las mismas propiedades intensivas en todos sus puntos.
Todo sistema homogéneo se caracteriza por presentar continuidad cuando se lo observa a simple vista, al microscopio y aún al ultramicroscopio. No es posible, en el ejemplo anterior, observar y distinguir el agua del azúcar.
Hay infinidad de sistemas homogéneos, entre otros: agua potable, aire (varios componentes); alcohol , agua (un componente), etc.

sistema homogéneo: agua pura
Sistemas Heterogéneos: si analizamos un sistema formado por agua y aceite (dos componentes), comprobamos que no posee homogeneidad, ya que a simple vista se distinguen la zona ocupada por el aceite y la zona ocupada por el agua. También podemos comprobar que ciertas propiedades intensivas (densidad por ejemplo) no se mantienen constantes cuando pasamos de un punto ocupado por el aceite a otro punto
ocupado por el agua. Lo mismo sucede en el sistema formado por agua líquida, hielo y vapor de agua -un componente-.
Los sistemas anteriores son heterogéneos y los podemos definir como: aquellos sistemas que presentan distintas propiedades intensivas en por lo menos dos de sus puntos.
Otros ejemplos de sistemas heterogéneos son: agua y arena, agua y limaduras de hierro, pólvora (clorato de potasio, carbono y azufre), etc.


sistemas heterogéneos
Homogeneidad y heterogeneidad son conceptos relativos que dependen de las condiciones experimentales. Sangre humana y leche son sistemas homogéneos a simple vista, pero observados con un microscopio revelan heterogeneidad; en la sangre se observan glóbulos rojos diferenciados del suero y en la leche gotitas de grasa. En consecuencia todo depende de como se ha practicado la determinación y que instrumento se ha empleado.
Dado que son numerosos los instrumentos utilizados: lupa, microscopio óptico común, microscopio electrónico, equipo de rayos X, etc., se ha convenido lo siguiente: los sistemas homogéneos y heterogéneos serán establecidos mediante el microscopio óptico habitual en laboratorios químicos y biológicos, con este aparato se visualizan hasta 10-4 cm (0,0001 cm).